Programs
We acquire and advance rare disease therapies that were deprioritized mid-development, ensuring they continue toward the patients who need them most.

Rugonersen
Angelman syndrome (AS) is a serious rare genetic neurodevelopmental disorder which causes severe mental and physical impairment and affects approximately 15,000 patients in each of the US and the EU5, with an estimated incidence of 1 in 12,000 to 20,000 live births (Mertz et al., 2013; Luk and Lo, 2016; Yakoreva et al., 2019).
AS is characterized by global developmental delay, intellectual disability, epilepsy (90% of cases before age 3 years) with an atypical underlying electroencephalogram (EEG), ataxia, tremor, hyperactivity, limited speech, and sleep dysregulation (Albrecht et al. 1997; Kishino et al. 1997; Knoll et al. 1989; Thibert et al. 2013). Symptoms often emerge during infancy and persist throughout life (Albrecht et al. 1997; Kishino et al. 1997; Knoll et al. 1989). Natural history (NH) studies have shown that AS is also characterized by EEG abnormalities that are particularly pronounced in the lower frequency range (𝛿-frequency-range, defined here as 2-4 Hz). In AS, EEG 𝛿-power has been shown to correlate cross-sectionally and longitudinally with symptom severity (Frohlich, J. et al. 2019, Hipp, J. F. et al 2021, Ostrowski, L. M. et al. 2021). Affected individuals require lifelong care and cannot live independently.
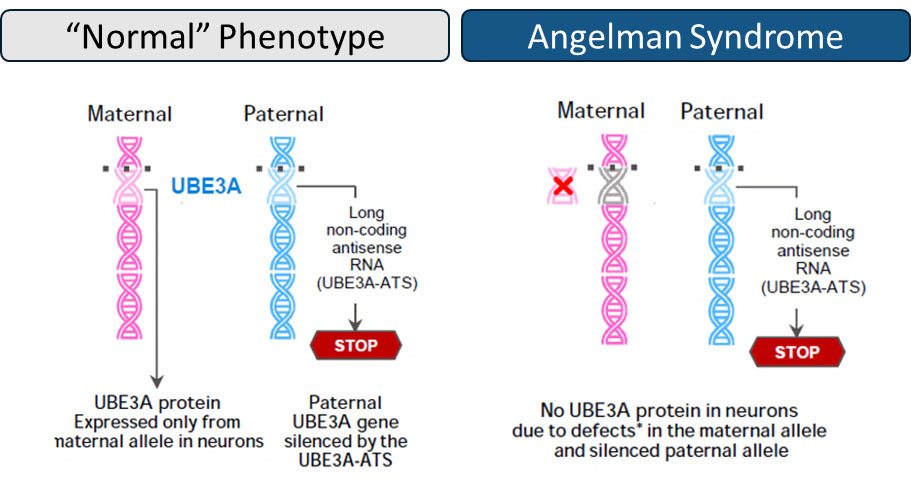
Deletions and mutations in the maternal ubiquitin protein ligase E3A (UBE3A) allele cause AS, as paternal UBE3A is epigenetically silenced by a long non-coding antisense RNA (UBE3A-ATS) in neurons. UBE3A is required for normal brain development and function. Failure to express ubiquitin E3A ligase in central nervous system (CNS) neurons leads to a build-up of damaged or unwanted proteins, that if left unchecked, can paralyze normal neuronal maturation, function, and synaptic pruning.
Rugonersen, an ASO designed to address the underlying disease biology of Angelman syndrome (AS). Rugonersen specifically and potently binds the UBE3A-ATS transcript. Rugonersen binding triggers degradation of the UBE3A-ATS transcript in the CNS and therefore the unsilencing of the UBE3A paternal allele. Rugonersen allows neuronal expression of the paternal wild-type copy of the UBE3A gene, potentially restoring normal neuronal function and development in AS patients.

In nonclinical studies, rugonersen demonstrates the ability to reduce UBE3A-ATS and increase UBE3A mRNA and protein (Jagasia et al, 2025). In the TANGELO study, an open-label, non-randomized, adaptive study led by Roche, rugonersen demonstrated encouraging exploratory effects compared to natural history on multiple clinical measures and on a pharmacodynamic biomarker of brain function, EEG delta power.
The clinical data are published in detail in a recent publication in Nature Medicine: https://www.nature.com/articles/s41591-025-03784-7
Hipp, J.F., Bacino, C.A., Bird, L.M. et al. The UBE3A-ATS antisense oligonucleotide rugonersen in children with Angelman syndrome: a phase 1 trial. Nat Med (2025). https://doi.org/10.1038/s41591-025-03784-7
Jagasia et al, Angelman syndrome patient-derived neuron screen leads to clinical ASO rugonersen targeting UBE3A-ATS with long-lasting effect in monkeys, Nucleic Acids Research (2025). https://doi.org/10.1093/nar/gkaf851
OHB-607
Every year, hundreds of thousands of infants worldwide are born extremely prematurely and, as a result, suffer from severe complications in their lungs, brain, and eyes. These complications can go on to hinder long-term development and quality of life.
While prenatal steroids, surfactants, ventilators and improved resuscitation protocols have increased the survival rate of premature infants, there has been little progress in protecting their not fully developed organs from the trauma of life-saving measures at birth, including supplemental oxygen and breathing machines.
There must be a strong commitment to looking for solutions for this vulnerable population.
OHB-607 has the potential to be the first breakthrough in more than 30 years to improve outcomes for these infants and their families.
Sources: 1 - Hellström A et al. Acta Paediatr 2016;105:576–586; 2 - Abman S et al Am J Resp Crit Care Med 2017,195:421-4; 3 - Ballabh P Pediatr Res 2010; 67: 1-8; 4 – Harnett ME


OHB-607 is a straightforward replacement therapy approach. OHB-607 (formerly TAK-607), is a proprietary, recombinant version of insulin-like growth factor 1 (IGF-1), the natural version of which is a key driver of fetal growth and development in utero, and its binding protein, IGFBP-3.
Mothers are the primary source of IGF-1 for the developing fetus, with the fetus producing very little of its own until reaching 30 weeks of gestational age. At birth, extremely premature infants, born at less than 28 weeks of gestational age, have low levels of IGF-1 which are associated with greater complication rates.* IGF-1 is crucial for organ development.
OHB-607, as a human IGF-1 replacement, is designed to make up for the deficiency that occurs in extremely preterm birth and thereby help promote continued development and maturation of vital organs and the vasculature that supports them.
OHB-607 has been evaluated in both preclinical and clinical studies. It has shown broad-based improvements in these preclinical studies. Additionally, in a Phase 2a clinical trial, OHB-607 showed a statistically significant improvement in preventing severe bronchopulmonary dysplasia and a positive trend in reducing intraventricular hemorrhage (pre-specified secondary endpoints), with no significant safety signal observed.*
*Hellström A, et al. Acta Paediatr 2016, 105: 576-586
*Ley D, et al. J Pediatr 2019;206:56–65.
OHB-607 is now in a Phase 2b study - current trial: Footprints
A Clinical Efficacy and Safety Study of OHB-607 in Preventing Bronchopulmonary Dysplasia in Extremely Premature Infants
https://clinicaltrials.gov/study/NCT03253263
Oak Hill Bio’s preclinical program brings exciting potential. OHB-401 leverages years of research in pKAL inhibition with a potential best-in-class oral small molecule for diabetic macular edema.
OHB-401
Diabetic macular edema occurs when excess fluid builds up around the macula due to leaky vessels and leading to swelling. DME is a leading cause of blindness.
In DME, the kallikrein system generates excess bradykinin, resulting in inflammation and edema in the retinal blood vessels. OHB-401 leverages years of research in pKAL inhibition with an oral small molecule for diabetic macular edema.
